
The Omicron coronavirus variant is likely the fastest-spreading virus in human history, according to experts. While one person with the measles virus—a standout among infectious microbes—might infect 15 others within 12 days, Omicron jumps from person to person so quickly that a single case can give rise to six cases after four days, 36 cases after eight days and 216 cases after 12 days. By mid-February, Omicron will infect up to 40 percent of the U.S. population, one projection estimates, dwarfing the 8 percent that get sick from flu each season.
When the Alpha variant was spotted in November 2020, scientists knew little about how its few mutations would affect its behavior. Now, with a year’s worth of knowledge and data, researchers have been able to link some of Omicron’s 50 or so mutations to mechanisms that have helped it spread so quickly and effectively. That investigative process normally takes a lot longer, says Sriram Subramaniam, a biochemist at the University of British Columbia. “But we’ve been looking at these variants for a year, so we were prepared,” he adds.
Omicron hosts twice as many mutations as other variants of concern, including 13 mutations on its spike protein that are rarely seen among other variants. Those changes to its anatomy have given it new and surprising abilities. If Delta is the brute-force Hulk variant, think of Omicron as the Flash—masked and wicked fast. Here, we explore four ways that the virus has physically changed. Three of those alterations have helped the virus evade our immune systems and become more infectious, while the fourth may have led it to produce more mild disease.
It donned a disguise. Scientists are reaching a consensus about what makes Omicron so transmissible, and most evidence points to a single, potent mechanism: among the variants, Omicron has an unparalleled ability to hide from the immune system.
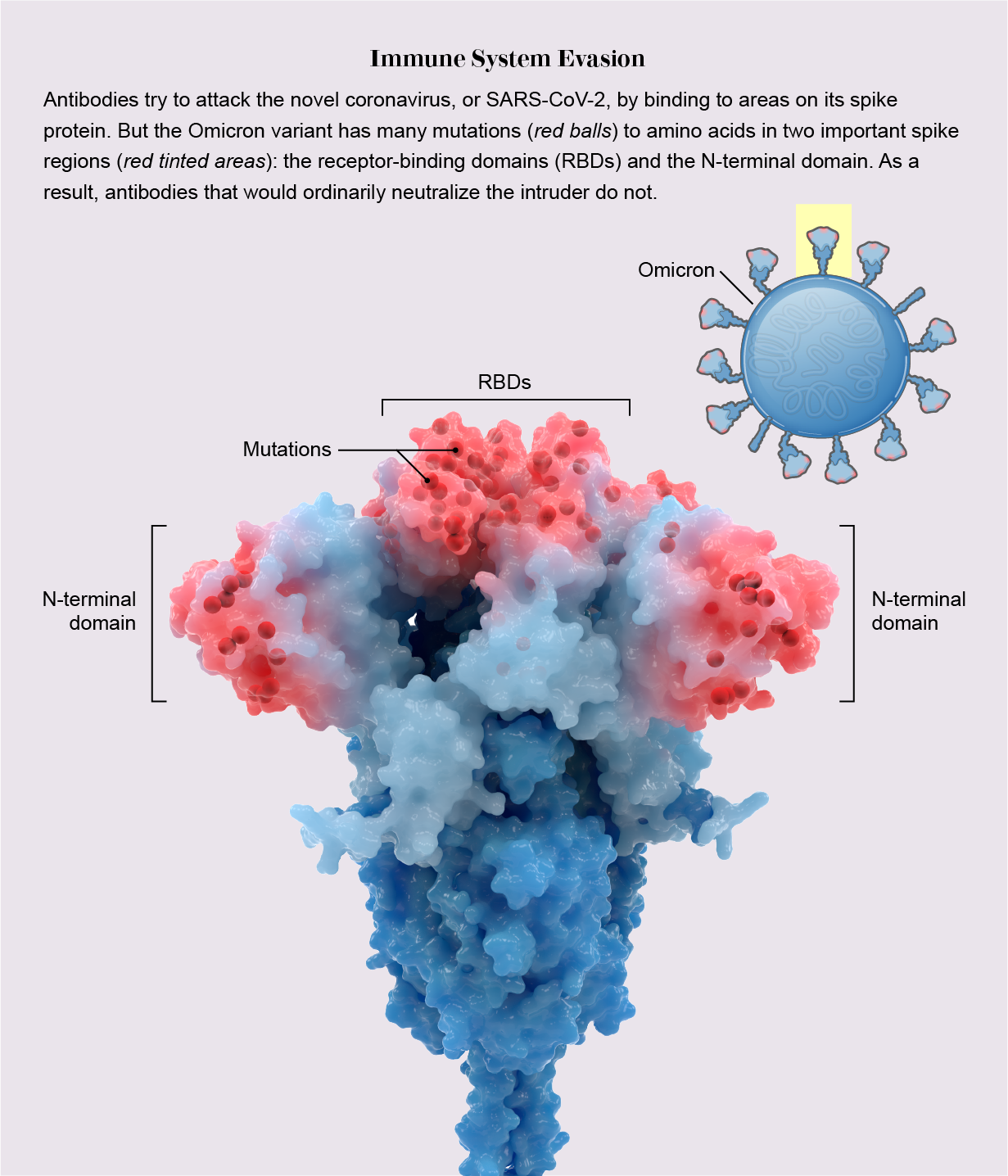
During infection, a fist-shaped clump of amino acids atop the coronavirus spike called receptor-binding domains (RBDs) grab onto a protein on the outside of human cells: the ACE2 receptor. To prevent that ill-fated attachment, the immune system creates antibodies—Y-shaped proteins induced by prior infection or vaccination—that recognize an RBD and stick to it like Velcro, getting in the way so the virus cannot link up with ACE2.
In previous variants, one, two or maybe three amino acids on RBDs were mutated, altering each RBD just enough to prevent some but not all antibodies from recognizing it. But Omicron harbors 15 RBD mutations, many on prime antibody-binding sites, forming an elaborate disguise to avoid many more antibodies. It is as if the virus donned a full-blown Mission: Impossible–style latex mask to change its face. “There are just so many mutations and so many new ones,” says Matthew McCallum, a biochemist at the University of Washington.
In a recent analysis published in the journal Science, McCallum, with his lab head David Veesler and their colleagues, showed a consequence of this dramatic transformation: only one of eight antibody treatments for COVID used in hospitals—which are based on natural antibodies—still bind to RBDs. Other research has shown mutations on RBDs and a second site called the N-terminal domain enable the virus to avoid antibodies gained by vaccination or infection. Thanks to Omicron’s convincing disguise, the variant has little to slow it down, and it spreads with lightning-fast speed. Vaccines, however, still ward off serious illness, especially with booster shots.
It stabilized. When Omicron drastically altered its spike to hide from the immune system, those changes eliminated some chemical residues the spike needed to attach to ACE2. But other mutations compensated: RBDs formed new chemical bridges to still effectively bind the protein, according to a recent study in Science. “It clearly lost some residues important for binding, but it made them up with other interactions,” says Subramaniam, who is lead author of the paper.
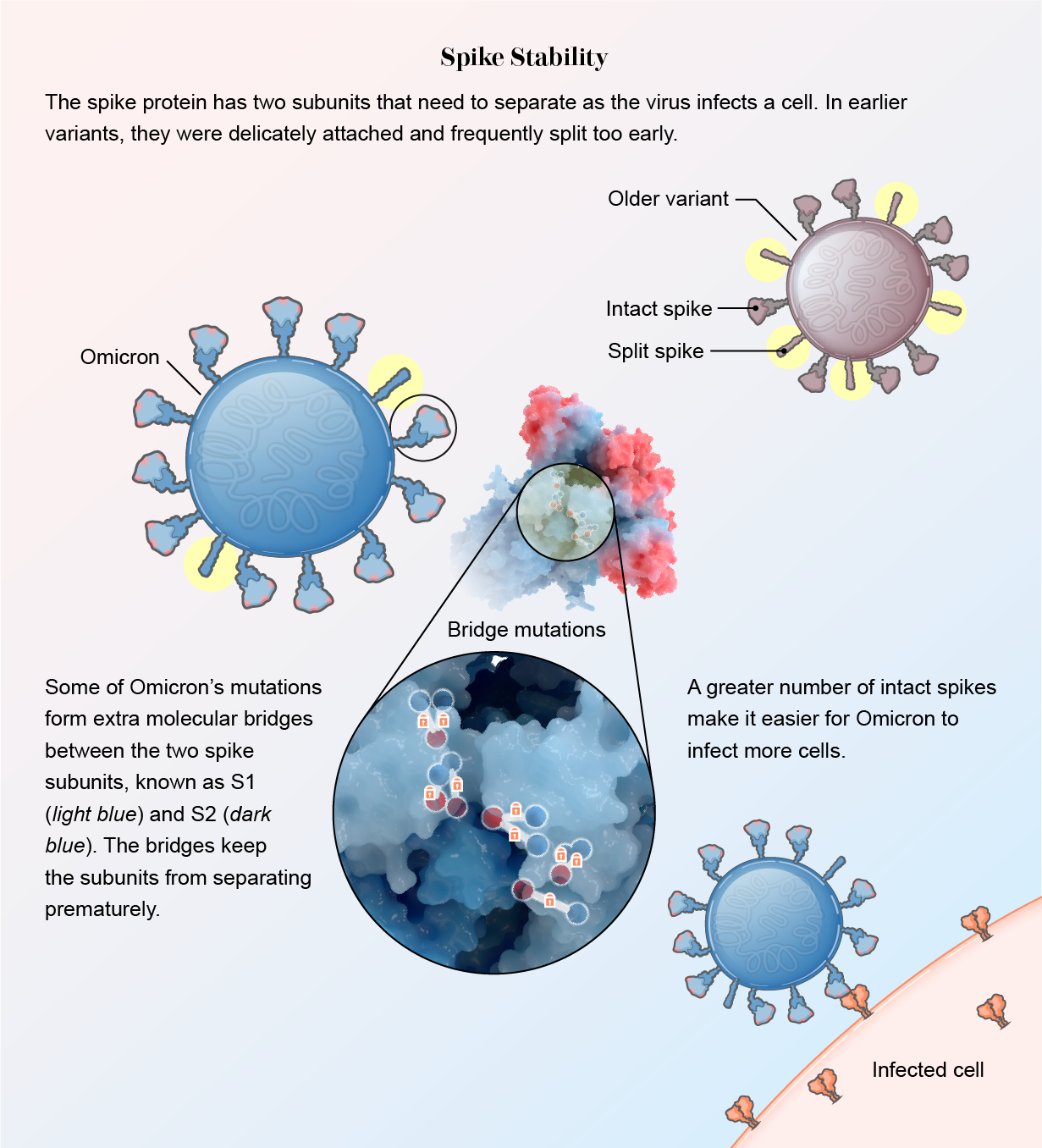
The spike protein also became sturdier. In other variants, two subunits within the spike, S1 and S2, are loosely connected. This allows them to split apart quickly so the spike can bury itself in a human cell when the virus encounters one. The downside of this delicate arrangement, however, is that many spikes split prematurely, before getting close to a cell. Once asunder, the spikes can no longer help the virus attach.
Mutations in Omicron have led to slim molecular bridges that hold the subunits together better, according to recent studies, one published in the Journal of Medical Virology and the others released as preprints, or studies that have not yet been formally reviewed by other scientists. “This virus has really protected itself from prematurely triggering,” says Shan-Lu Liu, author of one of the papers and director of the Viruses and Emerging Pathogens program at the Ohio State University. “When the virus is in the right place at the right time, it can be triggered and get into the cell, but not prior to that.”
It slipped in the side door. Across previous variants, there’s been one constant: The virus relies on a protein on the surface of human cells called TMPRSS2 (pronounced “tempress two”) to help it break through the cell membrane. But Omicron is not using TMPRSS2. It is taking a wholly different route into the cell. Instead of breaking down the front door, it slips in through the side.
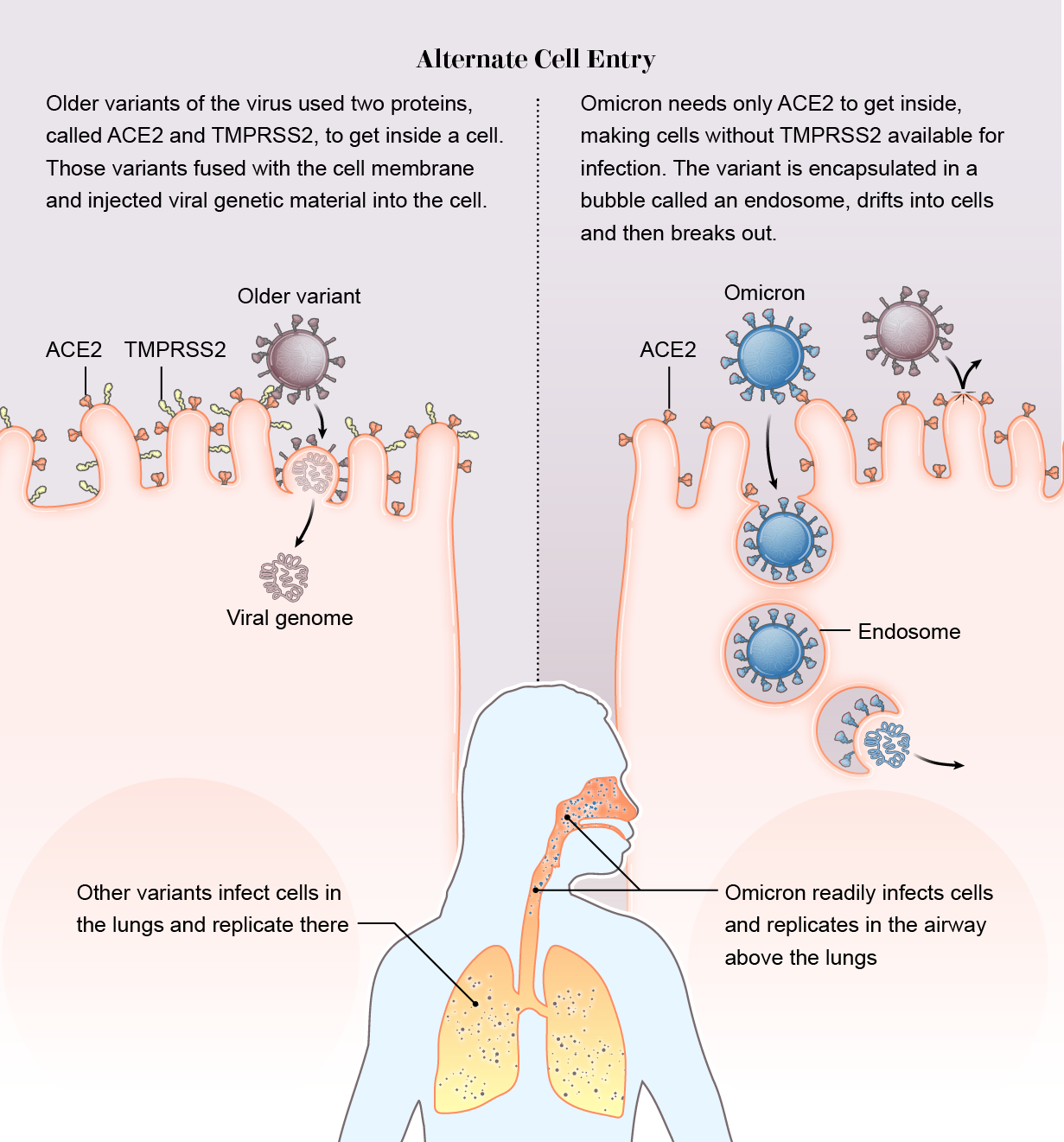
While other variants require both the ACE2 and TMPRSS2 proteins to inject their genome into a cell, Omicron binds only to ACE2. Then it is engulfed in a hollow bubble called an endosome. The bubble drifts into the cell, where the virus breaks out and begins a takeover.
Scientists speculate that Omicron gains two possible advantages this way. First, many cells do not have TMPRSS2 on their exterior, so if the virus does not need the surface protein, it has a wider buffet of cells to infect. “The current hypothesis is that there should be maybe seven or even 10 times more cells available to the virus if it goes through endosomes and isn’t reliant on TMPRSS2,” says Wendy Barclay, a virologist at Imperial College London, whose team, among others, detected the new entry pathway, which is described in a preprint.
Second, while the Delta variant often dives down to infect TMPRSS2-rich lung cells, Omicron replicates quickly in the airway above the lungs, which likely helps it spread from person to person. “We may be seeing a switch to the upper airway, which is promoting spread of the virus through coughing, sneezing and such,” says Joe Grove, a virologist at the University of Glasgow and co-author of a preprint that also detected the entry change.
It dropped its defenses. A final, fourth change to Omicron’s anatomy has not helped to make the variant more infectious, unlike the first three. Instead the alteration has created a surprising weakness, rendering the variant more vulnerable to a part of our bodily defenses known as the innate immune system.
Scientists examined the responses from Omicron and Delta to small proteins called interferons, which act like highway flares that alert innate immune cells to invaders. Delta was masterful at subduing the interferon response—but Omicron was terrible. It actually activated interferon signaling.
Researchers do not yet know how this change came about. At least 11 of the coronavirus’s 26 proteins interact with the interferon system, and many of those are mutated in Omicron. But even without knowing the exact mechanism, scientists can see hints of the consequences of this change.
Because the lungs are characterized by a more pronounced interferon response than the upper respiratory tract, Omicron’s vulnerability to that response may prevent it from spreading to the deeper organ. “It makes biological sense for what we see,” says Martin Michaelis, a biologist at the University of Kent in England, who analyzed how Omicron interacts with interferon in a paper published in Cell Research. “Omicron seems to be less capable of making it further into the body and into the lungs to cause severe disease.”
While Omicron’s impact on our whole population is not mild—including a giant surge in hospitalizations and deaths and a record number of hospitalized children—the new variant does appear to cause less severe disease in some infected individuals, as well as in animal models. Those who are unvaccinated or have other risk factors are still at greatly heightened risk for severe symptoms and death, however.
Additional mechanisms behind the variant’s unusual behavior are likely to be identified in coming months. “The picture always evolves over time,” Michaelis says. And future variants, when they appear, may have yet other modifications. “I’m not confident that we can rest on our laurels and say this is all over,” Barclay says. With infections continuing to spread and evolve among many populations around the world, the virus is going to come up with more ways to transmit—including ones that scientists haven’t even thought of yet.